Utente:Lorenzo Cecconi/Sandbox

- Nel mondo, nel 2002[1]
- Sono stati diagnosticati
- 1.352.100 nuovi casi di tumore del polmone
- Sono morte
- 1.178.900 persone per tumore del polmone
- Verranno diagnosticati
- 219.440 casi di tumore del polmone (116.090 negli uomini e 103.350 nelle donne)
- Moriranno
- 159.390 persone per tumore del polmone
- In Italia, ogni anno
Nel mondo
Il tumore del polmone è la neoplasia con il maggior tasso di incidenza e di mortalità nel mondo (1.35 milioni di nuovi casi all'anno e 1.18 milioni di morti), con la massima frequenza negli Stati uniti d'America e nell'Europa[1]. Vengono colpiti prevalentemente soggetti con età superiore a 50 anni e con una storia di fumo alle spalle. Le misure di prevenzione per il fumo di sigaretta prese dal 1960 in poi hanno portato a una lenta ma costante diminuzione del tasso di mortalità negli individui di sesso maschile nella prima parte di questo secolo[3], benché non si sia ancora osservata una diminuzione significativa nelle donne[4]. In particolare è stato rilevato che mentre nell'Europa dell'est il tasso di mortalità è maggiore negli uomini, nell'Europa del nord e negli Stati uniti d'America il tasso di mortalità è maggiore nelle donne[5]. Altri studi epidemiologici si sono concentrati nella valutazione degli altri fattori di rischio per lo sviluppo di tumore polmonari, rivelando un maggiore tasso di incidenza nelle popolazioni esposte all'inquinamento proveniente dalla emissioni di automobili, industrie e centrali termoelettriche, come nel Texas[6], in Taiwan[7] e nelle zone limitrofe a Dublino[8]. Dai dati provenienti da questi studi è risultato evidente il ruolo delle misure preventive focalizzate sulla riduzione dell'esposizione soprattutto ai fumi provenienti dalla combustione del gasolio e dei carburanti derivati dal petrolio[9].
Il tumore del polmone è meno comune nei paesi in via di sviluppo, benché è stato rilevato un notevole aumento di incidenza e di mortalità nei paesi in cui è subentrata l'abitudine al fumo di sigaretta, in particolare in Cina[10] e in India[11].
L'incidenza (per ogni paese) di neoplasie polmonari ha una relazione inversa con l'esposizione alla luce solare e ai raggi ultravioletti; una possibile spiegazione del fenomeno può essere connessa al ruolo anti-tumorale svolto dalla vitamina D che si origina dalla pelle in seguito all'esposizione solare[12].
Un dato degno di nota è un aumento dell'incidenza, dal 1950 in poi, della variante adenocarcinoma[13], tumore che interessa soprattutto le regioni periferiche del polmone. Il fenomeno è essenzialmente dovuto all'introduzione del filtro nelle sigarette, in grado di intrappolare le particelle più grandi (che si depositerebbero nei bronchi prossimali) e lasciar invece passare le particelle più piccole, che si depositano nei bronchi distali; non solo, la presenza di filtro porta il fumatore a fare inspirazioni più profonde per ricevere la stessa quota di nicotina, con maggiore deposizione delle sostanze tossiche nelle regioni polmonari più periferiche[14]. Tuttavia, negli Stati uniti d'America l'incidenza di adenocarcinoma sta diminuendo dal 1999; questo dato sempre essere dovuto alla diminuzione dell'inquinamento ambientale[13].
In Europa
| Nazione | Incidenza nei maschi | Incidenza nelle femmine |
|---|---|---|
In Italia


akgikhgbcsacasakgikhgbcsacasakgikhgbcsacasakgikhgbcsacasakgikhgbcsaca
sakgikhgbcsacasakgikhgbcsacasakgikhgbcsacasakgikhgbcsacasakgikhgbcsacasak
gikhgbcsacasakgikhgbcsacasakgikhgbcsacasakgikhgbcsacasakgikhgbcsacasakgikhgbcsac
asakgikhgbcsacasakgikhgbcsacasakgikhgbcsacasakgikhgbcsacasakgikhgbcsacasakgikhgbcsacasakgi
khgbcsacasakgikhgbcsacasakgikhgbcsacas
Già presente
Prevention is the most cost-effective means of fighting lung cancer. While in most countries industrial and domestic carcinogens have been identified and banned, tobacco smoking is still widespread. Eliminating tobacco smoking is a primary goal in the prevention of lung cancer, and smoking cessation is an important preventative tool in this process.[15] Most importantly, are prevention programs that target the youth. In 1998 the Master Settlement Agreement entitled 46 states in the USA to an annual payout from the tobacco companies.[16] Between the settlement money and tobacco taxes, each state's public health department funds their prevention programs, although none of the states are living up to the Center for Disease Control's recommended amount by spending 15 percent of tobacco taxes and settlement revenues on these prevention efforts.[16]
Policy interventions to decrease passive smoking in public areas such as restaurants and workplaces have become more common in many Western countries, with California taking a lead in banning smoking in public establishments in 1998. Ireland played a similar role in Europe in 2004, followed by Italy and Norway in 2005, Scotland as well as several others in 2006, England in 2007, and France in 2008. New Zealand has banned smoking in public places as of 2004. The state of Bhutan has had a complete smoking ban since 2005.[17] In many countries, pressure groups are campaigning for similar bans. In 2007, Chandigarh became the first city in India to become smoke-free. India introduced a total ban on smoking at public places on Oct 2 2008.
Arguments cited against such bans are criminalisation of smoking, increased risk of smuggling, and the risk that such a ban cannot be enforced.[18]
A 2008 study performed in over 75,000 middle-aged and elderly people demonstrated that the long-term use of supplemental multivitamins—such as vitamin C, vitamin E, and folate—did not reduce the risk of lung cancer. To the contrary, the study indicates that the long-term intake of high doses of vitamin E supplements may even increase the risk of lung cancer.[19]
The World Health Organization has called for governments to institute a total ban on tobacco advertising in order to prevent young people from taking up smoking. They assess that such bans have reduced tobacco consumption by 16% where already instituted.[20]
L'esposizione al fumo di sigaretta e agli altri fattori di rischio si traduce in modificazioni delle cellule dell'epitelio dei bronchi, soprattutto di quelli di primo, secondo e terzo ordine, da dove origina la maggior parte dei tumori polmonari[21]. Di questi, la maggioranza (90-95%) è rappresentata da tumori maligni di origine epiteliale (carcinomi) mentre solo il 5% dai carcinoidi bronchiali e il 2-5% da tumori mesenchimali[22]. Le forme epiteliali si distinguono in "tumori a piccole cellule" (SCLC) e "tumori non a piccole cellule" (NSCLC).
Classificazione istologica
| Classificazione istologica | Frequenza (%) |
|---|---|
| Carcinoma polmonare non a piccole cellule (NSCLC) | 80.4 |
| Carcinoma polmonare a piccole cellule (SCLC) | 16.8 |
| Carcinoide[24] | 0.8 |
| Sarcoma[25] | 0.1 |
| Altri istotipi | 1.9 |
| Sottotipi istologici di NSCLC |
Frequenza di NSCLC (%) | ||
|---|---|---|---|
| Fumatori | Non fumatori | ||
| Carcinoma a cellule squamose (carcinoma epidermoide) | 42 | 33 | |
| Adenocarcinoma | Adenocarcinoma (non altrimenti specificato) | 39 | 35 |
| Carcinoma bronchioloalveolare | 4 | 10 | |
| Carcinoide | 7 | 16 | |
| Altri | 8 | 6 | |
L'organizzazione mondiale della sanità prevede la divisione in 4 principali tipi istologici; tuttavia, ai fini terapeutici e prognostici è oppurtuno dividere i tumori del polmone in 2 grandi gruppi[27], definiti come carcinomi polmonari non a piccole cellule (NSCLC) e carcinoma polmonare a piccole cellule (SCLC). All'interno degli NSCLC si possono distinguere diverti sottotipi istologici, tra i quali
- Carcinoma a cellule squamose o carcinoma squamoso o carcinoma epidermoide
- Adenocarcinoma polmonare
- Carcinoma bronchioloalveolare, talvolta considerato come ulteriore sottotipo dell'adenocarcinoma bronchiale
- Carcinoma polmonare a grandi cellule
Il carcinoma epidermoide è un tumore maligno prevalentemente centrale che origina della cellule basali dell'epitelio dei bronchi, con lenta crescita e che predilige i lobi polmonari superiori; è la forma più strettamente associata al fumo di sigaretta[28]. L'adenocarcinoma del polmone è una forma tumorale estremamente variabile con linfotropismo estremamente marcato e in grado di formare metastasi linfonodali senza ingrandimento TAC visibile[29]. Il carcinoma bronchioloalveolare nasce dagli alveoli e diffonde tappezzando la parete alveolare, risultando non strettamente invasivo; ha un decorso clinico indolente e una frequenza molto bassa, con un quadro radiologico simile alla polmonite.
Il carcinoma polmonare a piccole cellule deve essere distinto da queste forme; è un tumore estremamente maligno, con elevatissima mortalità e che si sviluppa in un contesto patogenetico molecolare differenze rispetto ai NSCLC; può essere definito come l'estremo maligno della linea dei tumori che originano dalle cellule neuroendocrine (carcinoidi tipici e atipici, carcinoma polmonare a grandi cellule). La malignità tipica di questi tumori viene giustificata dal decorso clinico estremamente rapido, dalla precocissima capacità di metastatizzare e all'associazione con sindromi paraneoplastiche[30].
Aspetto microscopico
L'epitelio bronchiale è formato da un monostrato di cellule cilindriche ciliate frammiste a cellule caliciformi mucipare. Mentre queste ultime, insieme alle ghiandole della sottomucosa, sono deputate alla produzione di muco, le prime devono sostenere un'intensa attività di clearance (pulizia) dell'epitelio stesso, trasportando il muco prodotto verso l'alto attraverso il movimento ritmato delle ciglia. Tra queste cellule si interpongono cellule del sistema APUD, in grado di secernere un insieme di sostanze con proprietà vasoattive e con attività sulla muscolatura liscia bronchiale[31]. Sono inoltre presenti cellule indifferenziate che forniscono popolazioni cellulari sempre nuove all'epitelio bronchiale.
-
 Immagine istologica A
Immagine istologica A
Carcinoma squamocellulare; sono presenti cellule con citoplasma intensamente eosinofilo dalla quali protrudono spine che netrono in contatto con altre cellule. In alto a destra è inoltre possibile notare la presenza di notevole cheratinizzazione. -
 Immagine istologica B
Immagine istologica B
Adenocarcinoma a cellule chiare; le strutture cellulari si organizzano formando aggregati pseudo-ghiandolari. Le cellule appaiono chiare per la notevole presenza di muco. -
 Immagine istologica C1
Immagine istologica C1
Carcinoma bronchioloalveolare; le cellule neoplastiche tappezzano i setti interalveolari rispettandone la struttura. -
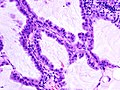 Immagine istologica C2
Immagine istologica C2
Carcinoma bronchioloalveolare, ulteriore immagine istologica. -
 Immagine istologica D
Immagine istologica D
Carcinoma polmonare a grandi cellule; sono presenti notevoli atipie nucleari, nucleolari e citoplasmatiche che rendono il quadro istologico notevolmente pleomorfo. Il nome dato alla neoplasia è giustificato dalla presenza di elementi cellulari giganti. -
 Immagine istologica E1
Immagine istologica E1
Carcinoma polmonare a piccole cellule; le cellule con nucleo intensamente cromofilo e scarso citoplasma si accrescono assumento una struttura compatta e dall'aspetto organoide. -
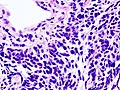 Immagine istologica E2
Immagine istologica E2
Carcinoma polmonare a piccole cellule, ulteriore immagine istologica.


.jpg)
.jpg)

_by_core_needle_biopsy.jpg)
_by_core_needle_biopsy.jpg)
-
 Rappresentazione A
Rappresentazione A
Carcinoma squamocellulare; sono presenti numerose spine tra cellula e cellula. Una perla cornea e parzialmente rappresentata nell'angolo in basso a destra. -
 Rappresentazione B
Rappresentazione B
Adenocarcinoma; la presenza di strutture ghiandolari più o meno differenziate testimonia la natura della neoformazione. -
Rappresentazione C
Carcinoma bronchioloalveolare; le cellule neoplastiche si accrescono tappezzando gli alveoli senza provocarne la distruzione. -
 Rappresentazione D
Rappresentazione D
Carcinoma polmonare a grandi cellule; il quadro è dominato da pleomorfismo marcato, con elementi cellulari giganti frammisti a cellule immature, con nucleoli prominenti e formazioni nucleari aberranti. -
 Rappresentazione E
Rappresentazione E
Carcinoma polmonare a piccole cellule; si può notare la scarsità del citoplasma e la presenza di nucleo intensamente cromofili. Le cellule si organizzano formando strutture compatte, dall'aspetto a palizzata.




- A - Carcinoma squamocellulare
- Come tutti i carcinomi a cellule squamose (o spinocellulare), questi tumori sono caratterizzati dalla presenza di cheratinizzazione, ben visibile per la presenza di elementi cellulari fusiformi intensamente eosinofili e totalmente o quasi totalmente privi di nucleo[32]. La combinazione e l'intensa proliferazione di questi elementi intorno ad un punto porta alla formazione di perle cornee, identificabili come zone concentriche con elevatissima cheratinizzazione (rappresentazione A, in basso a destra). Nell'immagine istologica A (e nella rappresentazione connessa) è inoltre possibile osservare la presenza di "spine" che protrudono dalla membrana cellulare e che formano ponti tra cellula e cellula. I ponti intercellulari altro non sono che desmosomi e testimoniano, unitamente alla cheratinizzazione, la conversione dell'epitelio cilindrico bronchiale in un epitelio molto più simile a quello della cute. La presenza di spine intercellulari è un reperto così caratteristico da poter render necessario la sua ricerca in ogni lesione bronchiale sospetta. La presenza di perle, di elementi squamosi e di spine delineano una forma di neoplasia ben differenziata; viceversa, l'assenza di perle cornee, la presenza di figure mitotiche atipiche e di elementi cellulari giganti ed immaturi possono far propendere più verso un reperto di neoplasia scarsamente differenziata, di con un grado maggiore di invasività[21].
- B - Adenocarcinoma
- Con il termine adenocarcinoma si fa riferimento ad una neoplasia di origine epiteliale in cui è possibile osservaere un certo grado di differenziazione verso l'epitelio ghiandolare, caratterizzato dalla presenza di struttutre acinose o papillari e dalla produzione di mucina[21]. Come mostra l'immagine istologica B, le cellule che si radudano a formare una struttura ghiandolare possono assumere una colorazione molto chiara per l'elevata presenza di mucina nel citosol. In questo caso si può parlare di adenocarcinoma a cellule chiare[32]. Invece, nella rappresentazione B si focalizza l'attenzione sulla formazione di strutture ghiandolari acinose che testimoniano il buon grado di differenziano del tumore; viceversa, ma mano che il tumore diventa più indifferenziato, le strutture acinari possono lasciare il posto a strutture papillari, che danno un aspetto più compatto al tessuto osservato. Con la progressiva perdita della differenziazione il tumore può assumere un aspetto solido, con elementi cellulari atipici e pleomorfi, di difficile caratterizzazione e inquadramento.
- C - Carcinoma bronchioloalveolare
- Questa variante istologica è sovente considerata una variante dell'adenocarcinoma, benché mostri una caratterizzazione anatomo-patologica, diagnostica e prognostica differente. Come è possibile vedere nelle immagini istologiche C1 e C2 e dalla rappresentazione C, le cellule tumorali proliferano rispettando la struttura microscopica polmonare, non invadendo la sottomucosa ma tappezzando come farfalle su di uno steccato (crescita lepidica) i setti alveolari. Le due varianti, muciparo e non muciparo, si differenziano per forma cellulare ed evoluzione; mentre i primi sono costituiti da cellule cilindriche e tendono a diffondersi per via aerogena, i secondi sono formati da cellule cuboidi o leggermente fusate, in grado di formare noduli spesso confinati[21].
- D - Carcinoma polmonare a grandi cellule
- Con questo termine si definisce una neoplasia polmonare altrimenti non identificabile nell'ambito di carcinoma squamocellulare o adenocarcinoma. Come suggerisce sia la rappresentazione che l'immagine istologica, questo tipo di tumore è formato da elementi cellulari pleomorfi, con nuclei prominenti e di dimensioni variabili dove è possibile apprezzare nucleoli intensamente cromofili (immagine istologica e rappresentazione D) . Questi tumori possono mostrare un certo grado di differenziazione neuroendocrina, confermata dall'aspetto di crescita organoide, con formazione di trabecole e rosette molto simili a quelle osservate nel microcitoma[33].
- E - Carcinoma polmonare a piccole cellule
- Questo tumore rappresenta l'estremo maligno di una serie di neoplasie che originano dalle cellule neuroendocrine, proprietà confermata dalla presenza di marcatori come la cromogranina o la sinaptofisina. Le cellule sono rotondeggianti, con scarso citoplasma e membrana cellulare ben definita; si organizzano formando strutture a palizzata (immagine istologica e rappresentazione E) con zone di necrosi e con elevatissima conta mitotica, fattore che testimonia il basso grado di differenziazione e l'elevata malignità[21].
Aspetto macroscopico
-
 Immagine A1
Immagine A1
Carcinoma squamocellulare, con caratteristica presentazione nelle zone centrali del polmone. -
 Immagine A2
Immagine A2
Carcinoma squamocellulare; il tumore si presenta come un'escrescenza verrucosa (a sinistra) occludende il bronco (destra) che si presenta eritematoso e contenente muco. -

-
 Immagine B
Immagine B
Adenocarcinoma polmonare; grande massa periferica, lobulata e dall'aspetto traslucido. -
 Immagine C
Immagine C
Carcinoma bronchioloalveolare; estese nodulazioni frammiste ad aree necrotico-emorragiche che alterano l'architettura polmonare. -
 Immagine D
Immagine D
Carcinoma polmonare a grandi cellule; estesa massa lobulata bianca e solida che presenta aree di necrosi focale. Il carcinoma si espande fino all'ilo polmonare dove sono inoltre presente linfonodi metastatici. -
 Immagine E
Immagine E
Carcinoma polmonare a piccole cellule; massa biancastra-cotonosa che avvolge un bronco di grosso calibro (sede ilare), infiltrando il tessuto peri-vascolare e intaccando i linfonodi ilari.







{kind=link}
- A - Carcinoma squamocellulare
- Sono molto spesso tumori centrali[32](immagine A1). Il carcinoma a cellule squamose si origina dall'epitelio bronchiale come un'escrescenza verrucosa (immagine A2) che può crescere fino ad occupare l'intero lume, provocando fenomeni di atelettasia o polmonite post-ostruttive, ben visibile nella figura (immagine A3). Il tumore può infiltrare la sottomucosa ed espandersi seguendo il tessuto peribronchiale in direzione mediastinica, dove appare radiograficamente come un estesa opacità in sede para-ilare (vicino all'ilo polmonare)[34]. Si presenta caratteristicamente come una massa "a cavolfiore", lobulata, di colore bianco-grigiastro e di consistenza dura; intorno alla massa possono presentarsi fenomeni necrotici (zone soffici e giallastre) ed emorragici (zone di polmone collassato infarcito di sangue)[21]. Talora, l'elevato tasso di crescita produce delle zone ischemico-necrotiche nella zona centrale che può escavarsi raccogliendo materiale purulento; in questi casi si parla di cancro-ascesso.
- B - Adenocarcinoma
- Sono spesso tumori che originano dai bronchi di piccolo calibro, benché possano anche presentarsi in sede parailare[33]. Assumono un aspetto lobulato e traslucido, in virtù della intensa produzione di mucina, come mostra l'immagine B.
- C - Carcinoma bronchioloalveolare
- La confluenza di più noduli (ben visibili nell'immagine C) porta alla formazione di un addensamento talmente esteso da poter essere confuso con una polmonite lobare. Talora sia presente un'intensa secrezione mucosa, la superficie di taglio può mostrarsi lucida, con aree più o meno grandi di raccolta<ref="mariu"/>. L'addensamento del parenchima, senza coinvolgimento bronchiale, porta allo sviluppo di broncogramma nei reperti radiografici, caratterizzato dalla visibilità delle strutture bornchiali contenti aria nel contesto del mezzo solido rappresentato dal tumore[29].
- D - Carcinoma polmonare a grandi cellule
- Questo tumore si sviluppa soprattutto nelle zone polmonari periferiche, benché, allo stesso modo dell'adenocarcinoma, possa estendersi in sede parailare[33]. Hanno un aspetto solido e lobulato (immagine D), possono assumere grandi dimensioni ed avere aree di raccolta ascessuale al loro interno.
- E - Carcinoma polmonare a piccole cellule
- Si sviluppano in sede ilare, coinvolgendo precocemente le strutture tracheali e mediastiniche[35]. Come mostra l'immagine E, il tumore tende ad infiltrare nel tessuto peribronchiale, coinvolgendo anche vasi sanguigni e linfonodi, benché possano presentarsi fenomeni di desquamazione necrotica intrabronchiale nel corso della malattia<ref="mariu"/>.
Alterazioni molecolari delle proteine o del DNA, unitamente allo stimolo irritativo, compongono il substrato necessario per la proliferazione cellulare e per la genesi di aberrazioni in senso metaplastico. Con il tempo e con il prolungarsi dell'esposizione, l'insieme di queste modificazioni costituisce il terreno sul quale origina e si muove la neoplasia. Da queste considerazioni si deduce che al disordine neoplastico si associano alterazioni molecolari, microscopiche e macroscopiche che evolvono nel tempo e nello spazio. Mentre il discorso sui disordini molecolari riguarda il profilo È stato già discusso di come alterazioni molecolari conducano una cellula verso Gli elementi eziologici analizzati portano bla bla bla... alterazioni dell'epitelio bla bla bla substrato molecolare sincrono ed evolutivo bla bla bla
Alterazioni clinico-patologiche
| Alterazioni istologiche nel tumore del polmone[21] | |
| Tappe anatomo-patogenetiche | |
| Stimolo oncogeno ↓ |
Stimolo oncogeno I I I I I ↓ Iperplasia adenomatosa atipica I I I I I ↓ |
| Alterazioni reversibili | |
| Iperplasia epiteliale ↓ Epitelio metaplastico ↓ | |
| Alterazioni irreversibili | |
| Displasia ↓ Carcinoma in situ ↓ | |
| Carcinoma polmonare a cellule squamose | Adenocarcinoma polmonare |
Come illustra lo schema (bla bla bla bla)
Eventi biologico-molecolari
Il terreno attraverso il quale si snoda la progressione bla bla bla ... bla bla .... alterazioni molecolari che possono essere riassunte in sei grandi gruppi patogenetici[36]
Autosufficienza per la crescita cellulare
Una cellula privata di fattori di crescita va rapidamente incontro ad apoptosi[37]. Analogamente, affiché un tessuto possa proliferare o andare incontro a differenziazione, occorono segnali extracellulari rappresentati da molecole di natura proteica (es: insulina, citochine), lipidica (es:cortisolo, triiodotironina) o peptidica; tali molecole vengono chiamate anche fattori di crescita o mitogeni. Affinché un fattore di crescita sia in grado di stimolare la proliferazione cellulare, occore che esso si leghi ad un recettore, localizzato nella membrana cellulare, nel citoplasma o nel nucleo. Il complesso fattore di crescita-recettore è in grado di attivare una cascata di segnalazione intracellulare il cui scopo finale è quello di promuovere la trascrizione di mRNA contenente le informazioni necessarie alla creazioni di proteine che innescano la divisione cellulare. Le tappe che dal complesso fattore di crescita-recettore portano alla divisioni cellulare sono molteplici; il motivo di tale complessità è quello di favorire e modulare il controllo di segnali che in maniera eccessivamente incauta possono promuovere una eccessiva proliferazione del tessuto. In questa maniera, per ogni scalino del processo di trasduzione, vengono attuati una serie di meccanismi di controllo il cui corretto funzionamento porta ad una corretta e coordinata proliferazione cellulare. Per esempio, questo meccanismo porta all'arresto della crescita in cellule poste ad un carico eccessivo di mitogeni, essendo magari una tappa a valle in grado di inibire a feedback una tappa a monte. Una cellula autosufficiente è una cellula che non ha bisogno della fisiologica quantità di stimoli esterni per provvedere alla crescita e al differenziamento. Diverse alterazioni, su diverse componenti, possono realizzare e promuovere l'autosufficienza: per il tumore del polmone assumono particolarmente importanza le vie di segnalazione cellulare che riguardano tre sistemi proteici: EGFR, Ras e Myc.

- EGFR
- Nel tessuto polmonare uno dei recettori per i fattori di crescita è rappresentato da EGFR (anche detto ErbB-1), proteina posta sulla membrana cellulare di molte cellule bronchiali[38].
Questo recettore è in grado di legare una proteina di 53 aminoacidi chiamata Epidermal growth factor (EGF) ma anche il Transforming growth factor α (TGFα)[39]; il legame con queste molecole porta all'attivazione del recettore, che omodimerizza con un altro recettore EGFR od eterodimerizza con altre proteine della famiglia dei recettori per il fattore di crescita epiteliale[38] come HER2/c-neu (ErbB-2), Her 3 (ErbB-3) e Her 4 (ErbB-4). Il partern più frequente di EGFR è rappresentato da HER2/c-neu[40]. La omo- eterodimerizzazione porta all'avvicinamento dei domini citoplasmatici dei suddetti recettori; tali domini possiedono un'attività tirosin chinasica, che innesca la vicendevole fosforilazione e successivo reclutamento di una serie di proteine come SOS[41], in grado di attivare, ad esempio Ras. In particolare, Ras è una delle principali proteine attivate dal legame dell'EGFR con il suo ligando[42]; in seguito alla sua attivazione, Ras diventa più affine per il GTP, perdendo viceversa la capacità di legare il GDP. Questa modificazione è tuttavia rapidamente reversibile, in quanto Ras, legandosi con GAP, diviene in grado di idrolizzare il GTP in GDP, perdendo nuovamente la sua attività. Per questo, l'attività di Ras viene definita pulsatoria. Tuttavia, nel breve periodo di attivazione, Ras è in grado di attivare la via delle Map chinasi, che porterà rapidamente alla differenziazione e proliferazione cellulare[43]. L'omo- eterodimerizzazione di EGFR non comporta soltanto l'attivazione di Ras tramite SOS, ma anche l'innesco della via di PI3K[44], il cui principale bersaglio è AKT[45] che, in seguito a attivazione, è in grado di inibire fortemente l'apoptosi. Un'ulteriore capacità del recettore EGFR è quella di attivare alcune proteine della famiglia STAT[40], che prendono parte all'innesco della proliferazione promuovendo la trascrizione di proteine come c-Myc. EGFR si configura dunque come un proto-oncogene a monte di una valle molto numerosa di possibili bersagli patogenetici la cui alterazione può portare all'autosufficienza dai fattori di crescita.
Essenzialmente, sono 3 i meccanismi attraverso i quali alterazioni di EGFR possono contribuire allo sviluppo e al mantenimento del cancro del polmone[38].
- Iperespressione dei ligandi di EGFR
- La continua presenza di molecole in grado di stimolare EGFR, benché non sia sufficiente ad innescare il processo di cancerogenesi[46], è estremamente importante poiché porta ad una condizione perpetua di stimolo proliferativo, promuovendo la proliferazione di cellule precedentemente mutate.
- Amplificazione di EGFR
- Se il numero di recettori per unità di superficie cellulare aumenta, aumenta di conseguenza la responsività della cellula ad un dato stimolo esterno[46]. Quindi, una cellula che esprime un maggior numero di recettori è una cellula con una maggiore capacità proliferativa, condizione che avvantaggia le cellule neoplastiche possiedono più copie del gene di EGFR o un gene costituitavamente espresso.
- Mutazioni attivanti di EGFR
- Le mutazioni del gene di EGFR possone portare all'indipendenza funzionale del recettore, rendendolo costitutivamente attivo anche in assenza di uno stimolo esterno[46]. Tali mutazioni sono presenti nel 20% dei NSCLC e nell'80% di NSCLC non responsivi a terapia[47], con maggiore frequenza nell'adenocarcinoma polmonare,nel sesso femminile e nei soggetti di origine asiatica[48].

- Ras
- Come precedentemente discusso nella trattazione del ruolo fisiologico di EGFR, la proteina Ras rappresenta un punto di snodo cruciale nella segnalazione di proliferazione e differenziazione cellulare[42]. Mutazioni di Ras, soprattutto dell'isoforma K-Ras, sono presenti soprattutto nell'adenocarcinoma polmonare[49], benché, comunque sia, rappresentano un'alterazione tipica (15-20%) di tutte le forme di NSCLC[50]. Mutazioni attivanti di K-Ras si associano molto strettamente all'abitudine al fumo di sigaretta e alla resistenza insorta durante il trattamento chemioterapico[51].
- Myc
- Myc è un oncogene che codifica per una proteina che rappresenta il traguardo finale del segnale di proliferazione convogliato da Ras[52]; ciò significa che una mutazione attivante di Myc o una sua iperespressione mima fisiologicamente una mutazione attivante di Ras. Le alterazioni di Myc sono associate a moltissime forme di cancro[53] e nel tumore del polmone assumono una valenza particolare le forme cMYC, MYCN e MYCL. Mentre la mutazione di cMyc in circa il 8-20% delle forme di NSCLS, la mutazione delle ultime due forme rappresenta un meccanismo patogenetico fondamentale nello sviluppo di SCLC[54].
Evasione dalla apoptosi
L'apoptosi può essere definita come morte cellulare programmata che, differentemente dalla necrosi, rappresenta un processo fisiologico e di notevolissima importanza. La regolazione dei processi apoptotici permette il corretta sviluppo di diversi nuovi tessuti a scapito di popolazioni cellulari senescenti o rudimentali. Benché questi processi sono particolarmente evidenti durante l'embriogenesi, l'apoptosi riveste un ruolo fondamentale anche nell'individuo adulto, soprattutto nella eliminazione dei linfociti autoreattivi e delle cellule tumorali[55]. Giocoforza, la compromissione di tali meccanismi o l'acquisizione da parte del tumore della capacità di evadere l'apoptosi, rappresenta uno dei momenti cruciali per la progressione della neoplasia[46].

- p53
- p53 è una proteina di 53 kilodalton (kDa) che funge da fattore di trascrizione ed è codificata dal gene TP53[56][57][58]. L'mRNA che traduce per p53 è trascritto in seguito a danno del genoma, provocato, ad esempio, da radiazioni, agenti chimici e stress ossidativo[59] . La proteina p53 cosi tradotta porta alla trascrizione di p21 (arresto del ciclo cellulare[60]), di GADD45 (riparazione del DNA[61][62] ) e di Bax (induttore dell'apoptosi[63]); riassumendo, un danno al DNA promuove la traduzione di p53 che blocca il ciclo cellulare, ripara il DNA e, in caso di insuccesso, innesca l'apoptosi. Per questo, p53 è stato denominato il guardiano del genoma, in quanto in grado di prevenire l'instaurarsi di danni al DNA e di stabilizzare il genoma[64]. Mutazioni inattivanti di p53 trasmesse con modalità autosomico recessiva sono le responsabili della sindrome di Li-Fraumeni[65], che rappresenta una condizione di rischio per lo sviluppo di cancro del polmone. Alterazioni di p53 nelle cellule dell'epitelio bronchiale con alterazioni di tipo neoplastico in individui che non hanno ereditato l'allele mutato, sono presenti sia nel microcitoma (>90%) che nei NSCLC (>80%)[27].
- Bcl-2
- Bcl-2 è una proteina che si lega alla membrana esterna dei mitocondri inibendo l'apoptosi[46]. Questo significa che un aumento dell'espressione di bcl-2 nelle cellule neoplastiche consente la valicazione degli stimoli apoptotici e la sopravvivenza cellulare. Benché l'iperespressione di bcl-2 rappresenti un punto patogenetico fondamentale nelle varie forme di leucemia e di linfomi, tale alterazione si riscontra frequentemente (>75%) anche nel SCLC[66]. In questo tipo di tumori è sovente riscontrare un'iperespressione di Bcl-2 e una diminuzione funzionale di p53, elementi che, con meccanismo sinergico, sono in grado di promuovere e sostenere l'aggressività del microcitoma.
Evasione dal blocco alla crescita cellulare
Nel processo di evoluzione di una neoplasia, le cellule acquisiscono gradualmente nuove capacità proliferative svincolandosi dal blocco imposto da alcuni geni denominati per questo oncosoppressori[46]. In generale, in meccanismi che portano alla perdita di un solo allele oncosoppressore non sono sufficienti a promuovere lo sviluppo di un tumore; tuttavia, la perdita di entrambi gli alleli (two hits hypothesis) è strettamente associata a instabilità genetica, ad alterato ciclo cellulare ed, infine, alla proliferazione incontrollata[67]. La proteina p53 rappresenta un tipico esempio di gene oncosoppressore; un ulteriore classico esempio è rappresentato dalla Rb, in grado di controllare le diverse tappe del ciclo cellulare[68]. Ogniqualvolta uno di questi geni viene perso o inattivato da mutazione in entrambi gli alleli si parla di loss-of-function[69] (LOH). Molti studi[70], focalizzati soprattutto sul carcinoma polmonare a cellule squamose, hanno messo in evidenza le seguenti loss-of-function
| Cromosoma coinvolto | Sigla | Esempi di geni presenti |
|---|---|---|
| Braccio corto del cromosoma 1 | 1p | PINK1 |
| Braccio corto del cromosoma 3 | 3p | FHIT; RASSF1; VHL |
| Braccio lungo del cromosoma 3 | 3q | PDCD10 |
| Braccio corto del cromosoma 4 | 4p | FGFR3 |
| Braccio lungo del cromosoma 4 | 4q | Molte chemochine |
| Braccio lungo del cromosoma 5 | 5q | NSD1 |
| Braccio lungo del cromosoma 8 | 8q | NDRG1 |
| Braccio lungo del cromosoma 9 | 9q | TGFBR1 |
| Braccio corto del cromosoma 10 | 10p | ERCC6 |
| Braccio lungo del cromosoma 10 | 10q | PTEN; ALOX5; CDH23 |
| Braccio lungo del cromosoma 13 | 13q | BRCA2; Rb |
| Braccio corto del cromosoma 17 | 17p | p53 |
| Braccio lungo del cromosoma 17 | 17q | BRCA1 |
| Braccio lungo del cromosoma 18 | 18q | SMAD4 |
| Braccio corto del cromosoma 19 | 19p | STK11 |
Recentissime ricerche[71] hanno messo in luce un ulteriore processo responsabile dell'evasione dal blocco alla crescita cellulare, rappresentato dalla metilazione dei promotori che modulano l'espressione di geni oncosoppressori. La metilazione è un processo fisiologico che porta al silenziamo di geni non necessari per la cellula nel dato periodo preso in considerazione[37]; tuttavia, la metilazione patologica che avviene in queste cellule tumorali può portare al silenziamento di oncosoppressori, con un profilo funzionale conseguente praticamente identico alla loss-of-function. Questi studi hanno inoltre messo in evidenza che tali alterazioni si verificano nelle fasi iniziali del processo di cancerogenesi, quando ancora non sono avvenute alterazioni mutazionali stabili. Ciò implica che le modificazioni a carico delle lesioni neoplastiche precoci possiedono ancora un certo grado di reversibilità.

Insensibilità all'invecchiamento cellulare
Nei laboratori di microbiologia l'identificazione di un virus avviene grazie all'incubazione in colture cellulari (piccoli contenitori rettangolari che contengono cellule prelevate da tessuti come il rene di scimmia) del materiale prelevato dal paziente sospetto[72]. Il più grosso limite di questa metodica è la scadenza cellulare, ovvero, il periodo oltre il quale una colonia di cellule non è più capace di sopravvivere anche se continuamente stimolata da fattori di crescita (vedi ^autosufficienza per la crescita cellulare). La scadenza imposta è comune a tutte le cellule dell'organismo umano; questo fenomeno, nell'insieme, è uno degli implicati nei processi di invecchiamento e senescenza dei tessuti e degli organi. Ad oggi, il problema della scadenza cellulare per le colture virali è stato valicato grazie alla presenza di colture in linea continua, gruppi cellulari che derivano da tumori, come le cellule HeLa (cellule dek carcinoma della cervice uterina di Henrietta Lacks). Le cellule in linea continua sono cellule immortalizzate (senza scadenza) da specifiche alterazioni a carico di alcuni geni, come quelli che codificano per la telomerasi[73], un enzima che stabilizza il genoma aggiungendo una specifica sequenza di DNA sull'estremità 3' di ogni cromosoma. L'insieme ripetuto delle sequenze specifiche aggiunte (TTAGGG, T = timidina, A = adenosina, G = guanosina) forma i telomeri, che costituisco il cappuccio terminale di ogni cromosoma. Con il ripetersi dei cicli cellulari e, dunque, delle mitosi, i telomeri si accorciano, testimoniando la fisiologica senescenza cellulare[37]. Da ciò si può dedurre che cellule con una spiccata attività telomerasi, quindi con telomeri costantemente lunghi, possono essere in grado di sfuggire ai normali processi di invecchiamento procedendo verso uno stato di immortalità. Nel tumore del polmone è stata ampiamente documentata[74] un'iperattività telomerasica soprattutto nelle fasi precoci del processo neoplastico. Presa visione di questa evidenza, numerosi studi[75][76] si sono focalizzati nel ricercare evidenze che possano suggerire il ruolo dell'esame dell'attività telomerasica sul liquido di lavaggio bronchiolo-alveolare (BAL) prelevato in corso di broncoscopia come misura di screening e prevenzione. Ulteriori studi[77] hanno analizzato la capacità predittiva dell'analisi dell'attività telomerasica associata alla normale citologia dell'espettorato o del BAL. Da queste ricerche risulta che, benché sia ancora una metodica in fase di sperimentazione e con costi elevati, l'analisi dell'attività telomerasica aggiunge sensibilità e accuratezza diagnostica al più specifico esame citologico, con ulteriore migliore capacità discriminativa sul grado di malignità del disordine neoplastico esaminato.

Nel corso dell'embriologia, ma anche nella vita adulta, lo sviluppo di tessuto o di un organo si accompagna ad un sincrono sviluppo dei vasi sanguigni da cui riceve ossigeno e sostanze nutritive. Il meccanismo alla base di questo processo deve essere ricercato nella secrezione da parte delle cellule del tessuto in espansione di una serie di fattori che stimolano la proliferazione dell'endotelio polarizzandolo verso il nuovo distretto. L'innesco alla secrezione di questi fattori di crescita è rappresentato dall'ipossia[46]; infatti, con l'espansione del tessuto, sempre più cellule si troveranno ad una distanza tale dal vaso sanguigno da risultare relativamente ischemiche. In queste cellule, soprattutto macrofagi tissutali, l'ischemia relativa porta ad una cascata di segnalazione intracellulare che ha come bersaglio la trascrizione di alcuni geni come il VEGF, il PDGF, l'EGF, l'FGF, il TGF-α e il TNF-α. Questo contesto di segnalazione molecolare, insieme alla presenza di proteasi e citochine, promuove lo sviluppo di nuovi vasi sanguigni, indirizzandoli verso le cellule relativamente ischemiche. Con il ritorno alla normale perfusione, lo stimolo proliferativo cessa e il nuovo tessuto è in grado di sopravvivere grazie al nuovo corredo vascolare. Dato che l'ossigeno diffonde nei tessuti per solo 1-2 mm rispetto al vaso sanguigno[78], un tumore che non possiede capacità angiogenetiche non può essere in grado di svilupparsi per uno spessore superiore a tale livello [79]. Viceversa, un tumore con spiccata attività angiogenica può procurarsi un corredo vascolare tale da sostenere una crescita virtualmente limitata. Non solo; con l'accesso al torrente ematico, un tumore può garantirsi il mezzo necessario alla propagazione metastatica a distanza. Numerosi studi sono stati condotti sul ruolo dei fattori di crescita per i vasi sanguigni nel tumore del polmone, dimostrando che il fattore cardine è rappresentato dal VEGF[80]; da ciò è stato reso evidente che un'elevata concentrazione di VEGF del sangue dei pazienti con tumore del polmone rappresenta un fattore prognostico negativo indipendente dalla stadiazione[81]. È stato inoltre dimostrato che il processo di angiogenesi sostenuto dal VEGF avviene nelle fasi precoci della cancerogenesi del tumore del polmone[82], con pesanti implicazioni nella storia naturale di queste neoplasie.

Invasione e metastizzazione
Una caratteristica che permette una prima distinzione tra tumori benigni e tumori maligni è il tipo di invasione tissutale; infatti, mentre i primi tendono ad espandersi comprimendo i tessuti circorstanti, i secondi tendono ad espandersi infiltrandoli[78]. Il processo di infiltrazione richiede la capacità attiva di farsi strada attraverso i tessuti, con demolizione della matrice extracellulare e sconvolgimento dell'archittettura dell'organo. Non solo: in un organo sano i vari tessuti che lo compongono sono in equilibrio stabile. Si consideri ad esempio un bronco, formato da un epitelio colonnare semplice che poggia in un esile strato connettivale che compone la lamina propria. La lamina propria divide la superficie epiteliale dalla sottomucosa, composta da connettivo nel quale sono immerse ghiandole sierose, tessuto muscolare liscio, fibre elastiche e cartilagine. L'architettura di un tessuto così complesso viene garantita dalla presenza di segnali di riconoscimento tra cellula e cellula, che impediscono lo sviluppo eccessivo dell'uno rispetto all'altro. Ad esempio, nella superficie laterale della membrana delle cellule dell'epitelio bronchiale sono presenti proteine chiamate caderine. La vicininanza tra cellule della stessa specie (es: epitelio) porta al contatto tra le varie caderine, che, così, innescano un segnale intracellulare che inibisce la proliferazione del tessuto. Questo meccanismo, chiamato inibizione da contatto, blocco lo sviluppo eccessivo di un tessuto e al contempo porta alla proliferazione dello stesso in caso di mancanza di cellule (es: una lesione). L'inibizione da contatto che si sviluppa tra cellule della stessa specie viene definita omotipica; quella che si sviluppa tra due cellule diverse (es: epitelio e tessuto connettivo) viene invece definita eterotipica, operante, ad esempio, nella parte inferiore dalla membrana cellulare delle cellule dell'epitelio bronchiale. Da ciò è possibile dedurre che il primo passo per l'infiltrazione è la perdita dell'inibizione da contatto. Alcuni studi hanno dimostrato che cellule epiteliali bronchiali possono mostrare una perdita dell'inibizione da contatto in seguito ad esposizione alla nicotina[83]. Il passo successivo alla perdita dell'inibizione da contatto è rappresentato dall'acquisizione della capacità di invasione tissutale attiva, grazie all'espressione di proteine, come le metallo proteasi, in grado di demolire la matrice extracellulare, permettendo alle cellule neoplastiche di farsi strada tra i tessuti. Molte di queste proteine sono chiamate in causa nella capacità invasiva del tumore del polmone; di seguito vengono discussi alcuni degli elementi ritenuti più importanti nell'infiltrazione di questa neoplasia.
- CRMP-1
- Collapsin response mediator protein 1 (proteina che media gli effetti delle collapsine) è un gene umano che codifica per una proteina di membrana presente fisiologicamente solo nel tessuto nervoso, dove modula vie di segnalazione importanti nella crescita neuronale durante lo sviluppo del sistema nervoso[84]. Al di là di questo ruolo fisiologico, CRMP-1 è stata indagata in quanto la sua espressione è alterata nelle cellule neoplastiche che possiedono un elevato grado di invasività[85]. Per quanto riguarda il tumore del polmone, studi condotti per valutare il ruolo del CTGF (fattore di crescita del tessuto connettivo, che stimola la produzione di matrice extracellulare e di integrine[86]) nella progressione dell'adenocarcinoma polmonare, hanno dimostrato una stretta relazione tra i segnali innescati da CRMP-1[87] e la motilità cellulare, con aumento del grado di invasione e di metastatizzazione. Non solo: dati precedenti a questi studi e basati su una serie di esperimenti condotti con la metodica PCR, northern blot e western blot[88], hanno dimostrato che cellule metastatiche e con elevato tasso di invasività presentano livelli ridotti di CRMP-1; da ciò è stato ipotizzato che CRMP-1 possa avere un ruolo anti-invasivo fisiologico.

- Laminina 5
- Le laminine sono proteine della matrice extracellulare, soprattutto dello strato compatto che costituisce la lamina propria che sostine le cellule epiteliali[89]. Sono formate da trimeri compositi di catene alfa, beta o gamma e le differenti combinazioni caratterizzano le diverse forme di laminine, espresse in differenti tessuti e con funzioni differenti[90]. Infatti, mentre una disfunzione della laminina 2 è responsabile di una forma di distrofia muscolare congenita, la laminina 1 è coinvolta nello sviluppo migratorio delle strutture neuronali, indirizzando, ad esempio, gli assoni delle cellule gangliari retiniche verso le stazioni mesencefaliche. Le laminine rappresentano dunque il terreno nel quale si sviluppano e si muovono le cellule; giocoforza, un'alterazione della produzione delle laminine può portare verso un'alterata architettura tissutale e nel caso di un tumore verso un certo grado di invasività. Alcuni studi hanno identificato nelle alterazioni della laminina 5 una tappa obbligata per l'invasità di molti tipi di tumori, di origine epiteliale e non[91]. Per il ruolo della laminina 5 nel tumore del polmone, l'attenzione è stata posta sull'adenocarcinoma in stadio Ia e Ib; le ricerche hanno dimostrato non solo che una bassa espressione della laminina 5 porta ad un grado di invasività minore, ma soprattutto correla con una prognosi a lungo tempo migliore[92].

- Integrine
- Le integrine sono recettori che mediano l'adesione tra le cellule e la matrice extracellulare[93]. In questo senso, sono responsabili della forma e della motilità cellulare, benché possiedano, analogamente a quando discusso per le caderine, proprietà segnalatorie in grado di influenzare il ciclo cellulare. Nello studio del ruolo delle integrine sullo sviluppo del tumore del polmono sono emersi i seguenti dati:
- L'invasività tissutale del carcinoma polmonare a cellule squamose, dell'adenocarcinoma polmonare, del carcinoma polmonare a grandi cellule e del carcinoma polmonare a piccole cellule è strettamente dipende dall'espressione nel microambiente polmonare di un contesto proteico che preveda la presenza di β1 e β3 integrine[94].
- L'espressione di proteine della matrice extracellulare come la β1 integrina è connessa con la resistenza alla chemioterapia e all'apoptosi mostrata in vivo dal carcinoma polmonare a piccole cellule[95].
- Molto recentemente è stato dimostrato che alcuni fattori di crescita come il TGF-β stimolano la produzione di integrine, promuovendo invasività e metastatizzazione delle cellule neoplastiche. In base a queste evidenze si stanno cercando nuovi farmaci in grado di bloccare questo processo[96].
- Ancor più recentemente (dati pubblicati nel giugno 2009) è stato dimostrato che la presenza di complessi recettoriali iperespressi formati da integrina β1 sia una conditio sine qua non affinché un cancro del polmone possa dar origine ai primi focolai metastatici[97].
Profilo clinico
Note
- ^ a b Incidence & Mortality, Lung cancer, su seer.cancer.gov, Surveillance, Epidemiology and End Results, 2009. URL consultato il 18 giugno 2009.
- ^ Incidence & Mortality, Lung cancer, su seer.cancer.gov, Surveillance, Epidemiology and End Results, 2009. URL consultato il 18 giugno 2009.
- ^ Alberg AJ, Samet JM., Epidemiology of lung cancer., in Chest, vol. 123, n. 1, January 2003, pp. 21S-41S.
- ^ Alberg AJ, Ford JG, Samet JM, Epidemiology of lung cancer: ACCP evidence-based clinical practice guidelines (2nd edition)., in Chest, vol. 132, n. 3, September 2007, pp. 29S-55S.
- ^ Gender in lung cancer and smoking research (PDF), su who.int, World Health Organization, 2004. URL consultato il 26 maggio 2007.
- ^ YM Coyle, Minahjuddin AT, Hynan LS, Minna JD, An ecological study of the association of metal air pollutants with lung cancer incidence in Texas., in Journal of Thoracic Oncology, vol. 1, n. 7, September 2006, pp. 654–661.
- ^ HF Chiu, Cheng MH, Tsai SS et al., Outdoor air pollution and female lung cancer in Taiwan., in Inhalation Toxicology, vol. 18, n. 13, December 2006, pp. 1025–1031, DOI:10.1080/08958370600904561.
- ^ Z Kabir, Bennett K, Clancy L, Lung cancer and urban air-pollution in dublin: a temporal association?, in Irish Medical Journal, vol. 100, n. 2, February 2007, pp. 367–369.
- ^ ME Parent, Rousseau MC, Boffetta P et al., Exposure to diesel and gasoline engine emissions and the risk of lung cancer, in American Journal of Epidemiology, vol. 165, n. 1, January 2007, pp. 53–62, DOI:10.1093/aje/kwj343.
- ^ BQ Liu, Peto R, Chen ZM et al., Emerging tobacco hazards in China: 1. Retrospective proportional mortality study of one million deaths, in British Medical Journal, vol. 317, n. 7170, 21 novembre 1998, pp. 1411–1422.
- ^ D Behera, Balamugesh T, Lung cancer in India (PDF), in Indian Journal of Chest Diseases and Allied Sciences, vol. 46, n. 4, 2004, pp. 269–281.
- ^ SB Mohr, Garland CF, Gorham ED et al., Could ultraviolet B irradiance and vitamin D be associated with lower incidence rates of lung cancer?, in Journal of Epidemiology and Community Health, vol. 62, n. 1, 2008, pp. 69–74, DOI:10.1136/jech.2006.052571.
- ^ a b F Chen, Bina WF, Cole P, Declining incidence rate of lung adenocarcinoma in the United States, in Chest, vol. 131, n. 4, April 2007, pp. 1000–1005, DOI:10.1378/chest.06-1695.
- ^ A Charloux, Quoix E, Wolkove N et al., The increasing incidence of lung adenocarcinoma: reality or artefact? A review of the epidemiology of lung adenocarcinoma, in International Journal of Epidemiology, vol. 26, n. 1, February 1997, pp. 14–23, DOI:10.1093/ije/26.1.14.
- ^ P Vineis, Hoek G, Krzyzanowski M et al., Lung cancers attributable to environmental tobacco smoke and air pollution in non-smokers in different European countries: a prospective study, in Environmental Health, vol. 6, BioMed Central, February 2007, p. 7, DOI:10.1186/1476-069X-6-7.
- ^ a b A Decade of Broken Promises: The 1998 State Tobacco Settlement Ten Years Later (PDF), su tobaccofreekids.org, Campaign for Tobacco-Free Kids. URL consultato il 3 dicembre 2008.
- ^ G Pandey, Bhutan's smokers face public ban, su news.bbc.co.uk, BBC, February 2005. URL consultato il 7 settembre 2007.
- ^ N Gray, A global approach to tobacco policy, in Lung Cancer, vol. 39, n. 2, BioMed Central, February 2003, pp. 113–117, DOI:10.1016/S0169-5002(02)00456-7.
- ^ Slatore CG, Littman AJ, Au DH, Satia JA, White E, Long-term use of supplemental multivitamins, vitamin C, vitamin E, and folate does not reduce the risk of lung cancer, in American Journal of Respiratory and Critical Care Medicine, vol. 177, n. 5, 2008, pp. 524–30, DOI:10.1164/rccm.200709-1398OC.
- ^ UN health agency calls for total ban on tobacco advertising to protect young, su un.org, United Nations News service, 30 May 2008.
- ^ a b c d e f g Robbins e Cotran, Le basi patologiche delle malattie - Volume 2 (7a edizione), Torino - Milano, Elsevier Masson, 2008, ISBN 9788885675537.
- ^ V. Colby, Tumors of the lower respiratory tract (3* ed), Washington DC, American Registry of Pathology, 1995, ISBN 1881041174.
- ^ WD Travis, Travis LB, Devesa SS, Lung cancer, in Cancer, vol. 75, Suppl. 1, January 1995, pp. 191–202.
- ^ U Morandi, Casali C, Rossi G, Bronchial typical carcinoid tumors, in Seminars in Thoracic and Cardiovascular Surgery, vol. 18, n. 3, 2006, pp. 191–198, DOI:10.1053/j.semtcvs.2006.08.005.
- ^ B Etienne-Mastroianni, Falchero L, Chalabreysse L et al., Primary sarcomas of the lung: a clinicopathologic study of 12 cases, in Lung Cancer, vol. 38, n. 3, December 2002, pp. 283–289, DOI:10.1016/S0169-5002(02)00303-3.
- ^ A Bryant, Cerfolio RJ, Differences in epidemiology, histology, and survival between cigarette smokers and never-smokers who develop non-small cell lung cancer, in Chest, vol. 132, n. 1, July 2007, pp. 198–192, DOI:10.1378/chest.07-0442.
- ^ a b Harrison, Principi di Medicina Interna (16a edizione), New York - Milano, McGraw-Hill, 2006, ISBN 88-386-2459-3.
- ^ Claudio Rugarli, Medicina interna sistematica (5a edizione), Masson, 2005, ISBN 9788821427923.
- ^ a b Giorgio Cittadini, Diagnostica per immagini e radioterapia, ECIG, 2008, ISBN 9788875441388.
- ^ Gianni Bonadonna, Gioacchino Robustelli Della Cuna, Pinuccia Valgussa, Medicina oncologica (8a edizione), Milano, Elsevier Masson, 2007, ISBN 978-88-214-2814-2.
- ^ Giuseppe C. Balboni, et al., Anatomia Umana, Vol. 1-2., Ristampa 2000, Milano, Edi. Ermes s.r.l., 1976, ISBN 8870510786.
- ^ a b c Mariuzzi, Anatomia patologica e correlazioni anatomo-cliniche, Padova, Piccin, 2006, ISBN 978882991769.
- ^ a b c James O'D. McGee, Peter G. Isaacson, Nicholas A. Wright, Heather M. Dick, Mary P. E. Slack, Oxford Textbook of Pathology, New York, Oxford University Press, USA, 2007, ISBN 978-0192619723.
- ^ Angelelli, AA.VV., Diagnostica per immagini per studenti e medici di medicina generale, Napoli, Idelson-Gnocchi, 2008, ISBN 978-88-7947-489-8.
- ^ Alberto Oliaro, Manuale di malattie dell'apparato respiratorio. Chirurgia toracica, pneumologia, Minerva medica, 2007, 9788877115478.
- ^ DeVita, Hellman, Lawrence, DeVita, Hellman, and Rosenberg's Cancer: Principles & Practice of Oncology (8a edizione)), Lippincott Williams & Wilkins, 2008, ISBN 9780781772075.
- ^ a b c Alberts, Johnson, Lewis, Raff, Roberts, Walter, Biologia molecolare della cellula (4a edizione), Bologna, Zanichelli, 2004, ISBN 8808078914.
- ^ a b c Zhang H, Berezov A, Wang Q, Zhang G, Drebin J, Murali R, Greene MI, ErbB receptors: from oncogenes to targeted cancer therapies, in J. Clin. Invest., vol. 117, n. 8, August 2007, pp. 2051–8, DOI:10.1172/JCI32278.
- ^ Luetteke NC, Lee DC, Transforming growth factor alpha: expression, regulation and biological action of its integral membrane precursor., in Semin. Cancer Biol., vol. 1, n. 4, 1991, pp. 265–75.
- ^ a b Olayioye MA, Update on HER-2 as a target for cancer therapy: intracellular signaling pathways of ErbB2/HER-2 and family members, in Breast Cancer Res, vol. 3, n. 6, 2001, pp. 385–389, DOI:10.1186/bcr327.
- ^ Nimnual A, Bar-Sagi D, The two hats of SOS, in Sci STKE, vol. 2002, n. 145, 2002, pp. PE36, DOI:10.1126/stke.2002.145.pe36.
- ^ a b Goodsell DS, The molecular perspective: the ras oncogene, in Oncologist, vol. 4, n. 3, 1999, pp. 263–4.
- ^ Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH, Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions, in Endocr. Rev., vol. 22, n. 2, 2001, pp. 153–83, DOI:10.1210/er.22.2.153.
- ^ Vanhaesebroeck B, Leevers S, Ahmadi K, Timms J, Katso R, Driscoll P, Woscholski R, Parker P, Waterfield M, Synthesis and function of 3-phosphorylated inositol lipids, in Annu Rev Biochem, vol. 70, 2001, pp. 535–602, DOI:10.1146/annurev.biochem.70.1.535.
- ^ Akt signaling pathway, in Product Pathways, Cell Signaling Technology, Inc.. URL consultato il 5 gennaio 2009.
- ^ a b c d e f g Pontieri - Russo - Frati, Patologia generale (3a edizione), Padova, Piccin, 2005, ISBN 88-299-1734-6.
- ^ Prudkin L, Wistuba II, Epidermal growth factor receptor abnormalities in lung cancer. Pathogenetic and clinical implications, in Annals of diagnostic pathology, vol. 10, n. 5, 2006, pp. 306–15, DOI:10.1016/j.anndiagpath.2006.06.011.
- ^ Ahmed SM, Salgia R, Epidermal growth factor receptor mutations and susceptibility to targeted therapy in lung cancer, in Respirology, vol. 11, n. 6, 2007, pp. 687–92, DOI:10.1111/j.1440-1843.2006.00887.x.
- ^ Bos J, ras oncogenes in human cancer: a review, in Cancer Res, vol. 49, n. 17, 1989, pp. 4682–9.
- ^ Downward J, Targeting RAS signalling pathways in cancer therapy, in Nat. Rev. Cancer, vol. 3, n. 1, January 2003, pp. 11–22, DOI:10.1038/nrc969.
- ^ Rotblat B, Ehrlich M, Haklai R, Kloog Y, The Ras inhibitor farnesylthiosalicylic acid (Salirasib) disrupts the spatiotemporal localization of active Ras: a potential treatment for cancer., in Methods Enzymol, vol. 439, 2008, pp. 467–89, DOI:10.1016/S0076-6879(07)00432-6.
- ^ Lüscher B, Function and regulation of the transcription factors of the Myc/Max/Mad network., in Gene, vol. 277, n. 1-2, 2001, pp. 1–14, DOI:10.1016/S0378-1119(01)00697-7.
- ^ Nilsson JA, Cleveland JL, Myc pathways provoking cell suicide and cancer., in Oncogene, vol. 22, n. 56, 2004, pp. 9007–21, DOI:10.1038/sj.onc.1207261.
- ^ Dang CV, O'donnell KA, Juopperi T, The great MYC escape in tumorigenesis., in Cancer Cell, vol. 8, n. 3, 2005, pp. 177–8, DOI:10.1016/j.ccr.2005.08.005.
- ^ Charles A. Janeway, Immunobiologia, Padova, Piccin, 2007, ISBN 8829918148.
- ^ Matlashewski G, Lamb P, Pim D, Peacock J, Crawford L, Benchimol S, Isolation and characterization of a human p53 cDNA clone: expression of the human p53 gene, in Embo J., vol. 3, n. 13, December 1984, pp. 3257–62.
- ^ Isobe M, Emanuel BS, Givol D, Oren M, Croce CM, Localization of gene for human p53 tumour antigen to band 17p13, in Nature, vol. 320, n. 6057, 1986, pp. 84–5, DOI:10.1038/320084a0.
- ^ Kern SE, Kinzler KW, Bruskin A, Jarosz D, Friedman P, Prives C, Vogelstein B, Identification of p53 as a sequence-specific DNA-binding protein, in Science (journal), vol. 252, n. 5013, June 1991, pp. 1708–11.
- ^ Han ES, Muller FL, Pérez VI, Qi W, Liang H, Xi L, Fu C, Doyle E, Hickey M, Cornell J, Epstein CJ, Roberts LJ, Van Remmen H, Richardson A, The in vivo gene expression signature of oxidative stress, in Physiol. Genomics, vol. 34, n. 1, June 2008, pp. 112–26, DOI:10.1152/physiolgenomics.00239.2007.
- ^ Rodriguez R, Meuth M, Chk1 and p21 cooperate to prevent apoptosis during DNA replication fork stress, in Mol. Biol. Cell, vol. 17, n. 1, January 2006, pp. 402–12, DOI:10.1091/mbc.E05-07-0594.
- ^ Fornace, A.J.; Jackman, J.; Hollander, M.C.; Hoffman-Liebermann, B.; & Liebermann, D.A., Genotoxic-stress-response genes and growth-arrest genes: gadd, MyD, and other genes induced by treatments eliciting growth arrest, in Annals of the New York Academy of Sciences, vol. 663, 1992, pp. 139–153.
- ^ Liebermann, D.A., Hoffman, B., Myeloid differentiation (MyD)/growth arrest DNA damage (GADD) genes in tumor suppression, immunity and inflammation, in Leukemia, vol. 16, n. 4, 2002, pp. 527–541, DOI:10.1038/sj.leu.2402477.
- ^ Z. N. Oltvai, Milliman, C. L. and Korsmeyer, S. J., Bcl-2 Heterodimerizes In Vivo with a Conserved Homolog, Bax, That Accelerates Programed Cell Death, in Cell, vol. 74, August 1993, pp. 609–619, DOI:10.1016/0092-8674(93)90509-O.
- ^ Read, A. P.; Strachan, T., Chapter 18: Cancer Genetics, in Human molecular genetics 2, New York, Wiley, 1999, ISBN 0-471-33061-2.
- ^ Giovanni Neri, Maurizio Genuardi, Genetica umana e medica, Milano, Elsevier Masson, 2008, ISBN 978-88-214-2917-0.
- ^ Tsung-Ming Yang, Dario Barbone, Dean A. Fennell e V. Courtney Broaddus, Bcl-2 Family Proteins Contribute to Apoptotic Resistance in Lung Cancer Multicellular Spheroids, in American Journal of Respiratory Cell and Molecular Biology, vol. 41, December 2008, pp. 14-23, DOI:10.1165/rcmb.2008-0320OC.
- ^ http://www.nature.com/scitable/topicpage/Tumor-Suppressor-TS-Genes-and-the-Two-887 Articolo sui geni oncosoppressori, sulla loro trasmissione autosomico recessiva e sulla two hits hypothesis nel sito nature.com
- ^ Murphree AL, Benedict WF, Retinoblastoma: clues to human oncogenesis, in Science, vol. 223, n. 4640, March 1984, pp. 1028–33, DOI:10.1126/science.6320372.
- ^ http://www.unisr.it/BiotechBook/view.asp?id=303 Definizione di LOH sul sito dell'Università Vita-Salute San Raffaele
- ^ S. Petersen, M. Aninat-Meyer, K. Schlüns, K. Gellert, M. Dietel, I. Petersen, Chromosomal alterations in the clonal evolution to the metastatic stage of squamous cell carcinomas of the lung, in British Journal of Cancer, vol. 82, December 1999, pp. 65-73, DOI:10.1054/bjoc.1999.0878.
- ^ Malcolm Brock, Craig M. Hooker, M.P.H., Emi Ota-Machida, M.D., Ph.D., Yu Han, B.S., Mingzhou Guo, M.D., Ph.D., Stephen Ames, B.A., Sabine Glöckner, M.D., Ph.D., Steven Piantadosi, M.D., Ph.D., Edward Gabrielson, M.D., Genevieve Pridham, B.S., Kristen Pelosky, B.S., Steven A. Belinsky, Ph.D., Stephen C. Yang, M.D., Stephen B. Baylin, M.D., James G. Herman, M.D., DNA Methylation Markers and Early Recurrence in Stage I Lung Cancer, in The New England journal of medicine, vol. 358, March 2008, pp. 1118-1128, DOI:10.1054/bjoc.1999.0878.
- ^ (IT) Anna M. Molina Romanzi, Giuseppe A. Botta; Letizia Calegari; Carlo Chezzi; Eugenio A. Debbia; Giorgio Palù; Gianni Pozzi; Carla Pruzzo; Luigi Robert; Gian Carlo Schito; Marco Toni; Maria Antonietta Tufano; Pier Egisto Valensin; Oliviero E. Varnier, Microbiologia clinica, Ristampa 2004, Torino, UTET, 2002, ISBN 8879332511.
- ^ Scott B. Cohen, Mark E. Graham, George O. Lovrecz, Nicolai Bache, Phillip J. Robinson, Roger R. Reddel, Protein composition of catalytically active human telomerase from immortal cells, in Science, vol. 315, 2007, p. 1850, DOI:10.1126/science.1138596.
- ^ http://cme.medscape.com/viewarticle/419199 Articolo su medscape.com su telomerasi, telomeri e connessione con il tumore del polmone
- ^ Yahata N, Ohyashiki K, Ohyashiki JH, Iwama H, Hayashi S, Ando K, Hirano T, Tsuchida T, Kato H, Shay JW, Toyama K., Telomerase activity in lung cancer cells obtained from bronchial washings., in Journal of national cancer institute, 90(9), 1998.
- ^ Kakihana M, Yahata N, Hirano T, Honda H, Ikeda N, Kawate N, Konaka C, Ebihara Y, Ohyashiki K, Kato H., Telomerase activity during carcinogenesis in the bronchus., in Oncology reports, 9(1), 2002.
- ^ Erkan Dikmena, Murat Karaa, Günnur Dikmenb, Hüseyin Çakmakc, Pakize Doganb, Detection of telomerase activity in bronchial lavage as an adjunct to cytological diagnosis in lung cancer, in European journal of cardio-thoracic surgery, vol. 23, november 2002, pp. 194-199.
- ^ a b Robbins e Cotran, Le basi patologiche delle malattie - Volume 1 (7a edizione), Torino - Milano, Elsevier Masson, 2008, ISBN 9788885675537.
- ^ Folkman J., The role of angiogenesis in tumor growth., in Seminars in cancer biology, 3(2), April 1992, pp. 65-71.
- ^ http://www.researchvegf.com/researchvegf/tumors/lung/index.m Pagina principale sul tumore del polmone nel sito researchvegf.com che raccoglie tutte le ricerche e i progressi scientifici sullo studio del VEGF
- ^ D. Brattströma, M. Bergqvista, P. Hesseliusa, A. Larssonb, G. Wageniusa, O. Brodinc, Serum VEGF and bFGF adds prognostic information in patients with normal platelet counts when sampled before, during and after treatment for locally advanced non-small cell lung cancer, in Lung cancer, 43(1), January 2004, pp. 55-62.
- ^ Adi F. Gazdar e John D. Minna, Angiogenesis and the Multistage Development of Lung Cancers., in Clinical cancer research, vol. 6, May 2000, pp. 1611-1612.
- ^ John D. Minna, Nicotine exposure and bronchial epithelial cell nicotinic acetylcholine receptor expression in the pathogenesis of lung cancer, in Journal of clinical investigation, 111(1), January 2003, pp. 31-33, DOI:10.1172/JCI17492..
- ^ Entrez Gene: CRMP1 collapsin response mediator protein 1, su ncbi.nlm.nih.gov.
- ^ Shih JY, Lee YC, Yang SC, et al., Collapsin response mediator protein-1: a novel invasion-suppressor gene., in Clin. Exp. Metastasis, vol. 20, n. 1, 2003, pp. 69–76, DOI:10.1023/A:1022598604565.
- ^ Brigstock DR (2002) Regulation of angiogenesis and endothelial cell function by connective tissue growth factor (CTGF) and cysteine-rich 61 (CYR61). Angiogenesis 5:153-165.
- ^ Chang CC, Shih JY, Jeng YM, et al., Connective tissue growth factor and its role in lung adenocarcinoma invasion and metastasis., in J. Natl. Cancer Inst., vol. 96, n. 5, 2004, pp. 364–75.
- ^ Shih JY, Yang SC, Hong TM, Yuan A, Chen JJ, Yu CJ, Chang YL, Lee YC, Peck K, Wu CW, Yang PC.', Collapsin response mediator protein-1 and the invasion and metastasis of cancer cells., in Journal of the national cancer institute, vol. 93, n. 18, september 2000, pp. 1392–1400.
- ^ M. A. Haralson and John R. Hassell, Extracellular matrix: a practical approach, Ithaca, N.Y, IRL Press, 1995, ISBN 0-19-963220-0.
- ^ Colognato H, Yurchenco P, <213::AID-DVDY1>3.0.CO;2-R Form and function: the laminin family of heterotrimers, in Dev. Dyn., vol. 218, n. 2, 2000, pp. 213–34, DOI:10.1002/(SICI)1097-0177(200006)218:2<213::AID-DVDY1>3.0.CO;2-R.
- ^ Charles Pyke, Sirpa Salo, Elisabeth Ralfkiær, John Rømer, Keld Danø and Karl Tryggvason, Laminin-5 Is a Marker of Invading Cancer Cells in Some Human Carcinomas and Is Coexpressed with the Receptor for Urokinase Plasminogen Activator in Budding Cancer Cells in Colon Adenocarcinomas1, in American association for cancer research, vol. 55, september 1995, pp. 4132-4139.
- ^ Moriya Y, Niki T, Yamada T, Matsuno Y, Kondo H, Hirohashi S., Increased expression of laminin-5 and its prognostic significance in lung adenocarcinomas of small size. An immunohistochemical analysis of 102 cases., in Cancer, vol. 91, n. 6, march 2001, pp. 1129-41.
- ^ Humphries M.J., Integrin structure, in Biochem. Soc. Trans., vol. 28, n. 4, 2000, pp. 311–339, DOI:10.1042/0300-5127:0280311.
- ^ CG Bredin, KG Sundqvist, D Hauzenberger, and J Klominek ., Integrin dependent migration of lung cancer cells to extracellular matrix components, in European respiratory journal, vol. 11, January 1998, pp. 400-407.
- ^ Tariq Sethi, Robert C. Rintoul, Sarah M. Moore1, Alison C. MacKinnon, Donald Salter, Chin Choo, Edwin R. Chilvers, Ian Dransfield, Seamas C. Donnelly, Robert Strieter, Christopher Haslett1, Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: A mechanism for small cell lung cancer growth and drug resistance in vivo, in Nature medicine, vol. 5, 1999, pp. 662-668, DOI:10.1038/9511.
- ^ Fong YC, Hsu SF, Wu CL, Li TM, Kao ST, Tsai FJ, Chen WC, Liu SC, Wu CM, Tang CH, Transforming growth factor-beta1 increases cell migration and beta1 integrin up-regulation in human lung cancer cells., in Lung cancer, vol. 64, n. 1, April 2009, pp. 13-21.
- ^ Shibue T, Weinberg RA., Integrin beta 1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs., in The Proceedings of the National Academy of Sciences (US), June 2009.
Cecco, ricorda che
Dimerizzazione (egfr) = appaiamento con altre proteine di membrana
Bibliografia
Oncologia
- DeVita, Hellman, Lawrence, DeVita, Hellman, and Rosenberg's Cancer: Principles & Practice of Oncology (8a edizione), Lippincott Williams & Wilkins, 2008, ISBN 9780781772075.
- Gianni Bonadonna, Gioacchino Robustelli Della Cuna, Pinuccia Valgussa, Medicina oncologica (8a edizione), Milano, Elsevier Masson, 2007, ISBN 978-88-214-2814-2.
Patogenesi e genetica
- Pontieri - Russo - Frati, Patologia generale (3a edizione), Padova, Piccin, 2005, ISBN 88-299-1734-6.
- Giovanni Neri, Maurizio Genuardi, Genetica umana e medica, Milano, Elsevier Masson, 2008, ISBN 978-88-214-2917-0.
Anatomia patologica
- Robbins e Cotran, Le basi patologiche delle malattie (7a edizione), Torino - Milano, Elsevier Masson, 2008, ISBN 9788885675537.
- Mariuzzi, Anatomia patologica e correlazioni anatomo-cliniche, Padova, Piccin, 2006, ISBN 978882991769.
- James O'D. McGee, Peter G. Isaacson, Nicholas A. Wright, Heather M. Dick, Mary P. E. Slack, Oxford Textbook of Pathology, New York, Oxford University Press, USA, 2007, ISBN 978-0192619723.
- V. Colby, Tumors of the lower respiratory tract (3* ed), Washington DC, American Registry of Pathology, 1995, ISBN 1881041174.
Medicina interna
- Harrison, Principi di Medicina Interna (il manuale - 16a edizione), New York - Milano, McGraw-Hill, 2006, ISBN 88-386-2459-3.
- Claudio Rugarli, Medicina interna sistematica (5a edizione), Masson, 2005, ISBN 9788821427923.
Diagnostica per immagini e radioterapia
- Fraser, Colman, Müller, Paré, Malattie del torace. Diagnostica per immagini e valutazione clinica (3a edizione), Milano, Elsevier Masson, 2006, ISBN 8885675875.
- Giorgio Cittadini, Diagnostica per immagini e radioterapia, ECIG, 2008, ISBN 9788875441388.
- Angelelli, AA.VV., Diagnostica per immagini per studenti e medici di medicina generale, Napoli, Idelson-Gnocchi, 2008, ISBN 978-88-7947-489-8.
Chirurgia
- Patterson, Cooper, Deslauriers, Griffith Pearson, Luketich, Pearson's Thoracic and Esophageal Surgery (3a edizione), Elsevier, 2008, ISBN 9780443068614.
- Alberto Oliaro, Manuale di malattie dell'apparato respiratorio. Chirurgia toracica, pneumologia, Minerva medica, 2007, 9788877115478.
- Dionigi, Basi teoriche e Chirurgia generale - Chirurgia specialistica (4° edizione), Padova, Elsevier Masson, 2006, ISBN 9788829916542.
- Mazzeo - Forestieri, Trattato di chirurgia oncologica, PICCIN - Nuova libreria, 2006, ISBN 8821429121.
Terapia farmacologica, Chemioterapia
- Brunton, Lazo, Parker, Goodman & Gilman - Le basi farmacologiche della terapia 11/ed, McGraw Hill, 2006, ISBN 788838639111.
- Bertram G. Katzung, Farmacologia generale e clinica, Padova, Piccin, 2006, ISBN 88-299-1804-0.